|
|
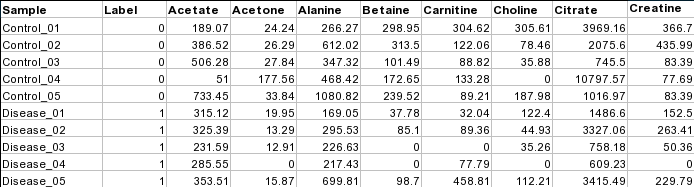
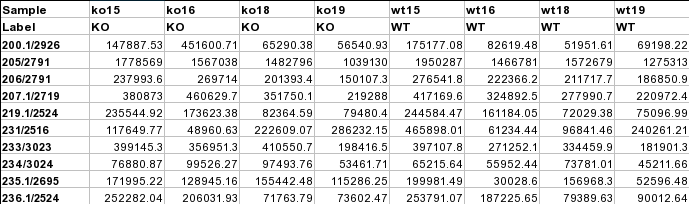
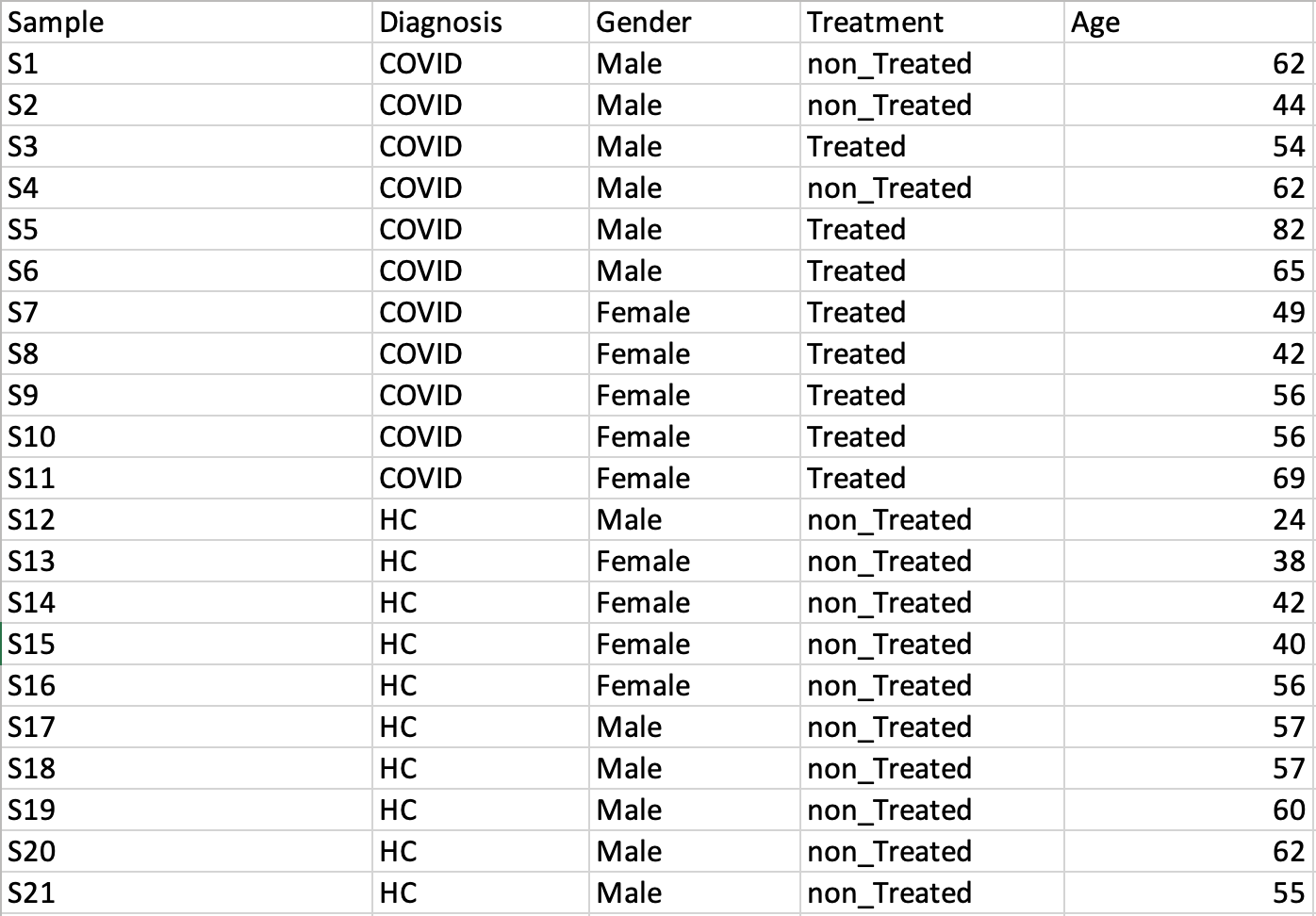
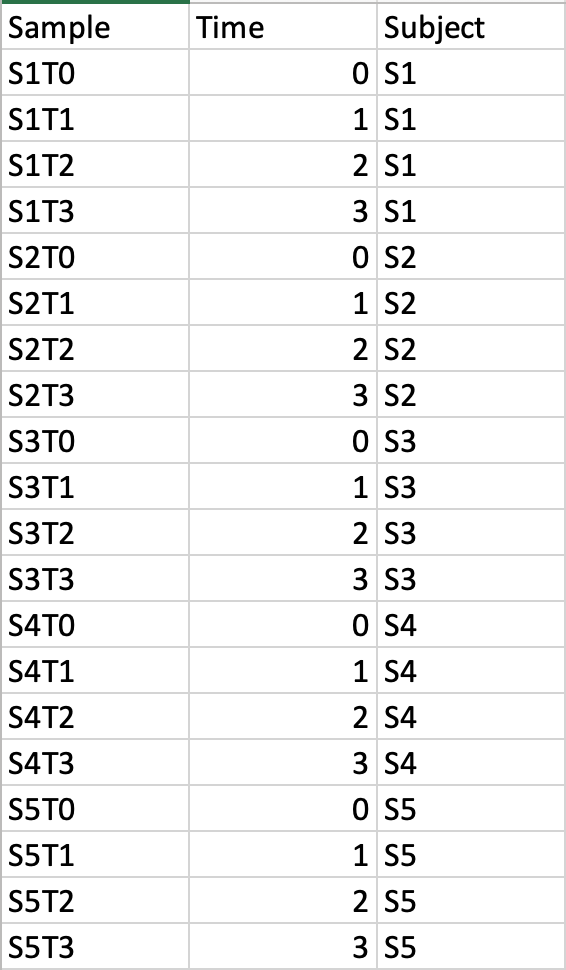
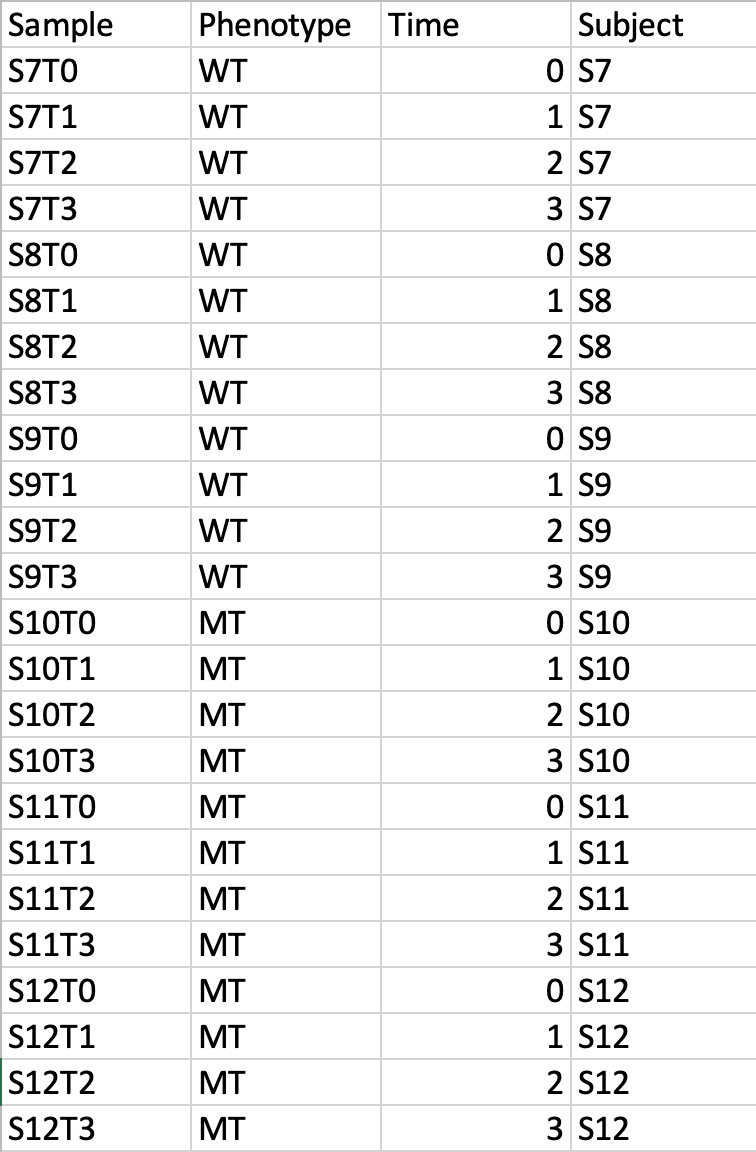
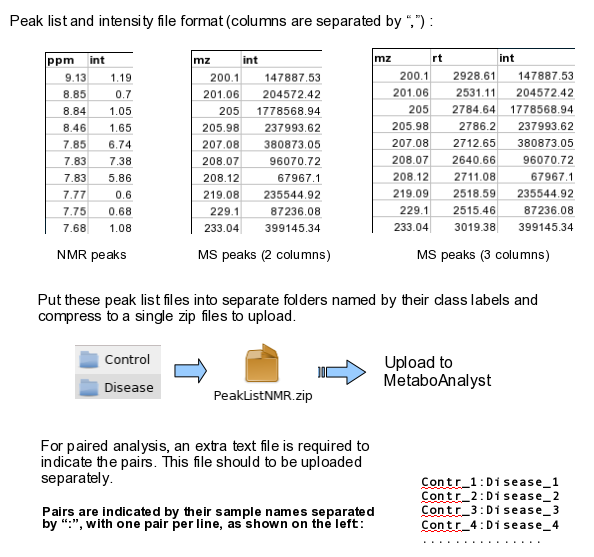
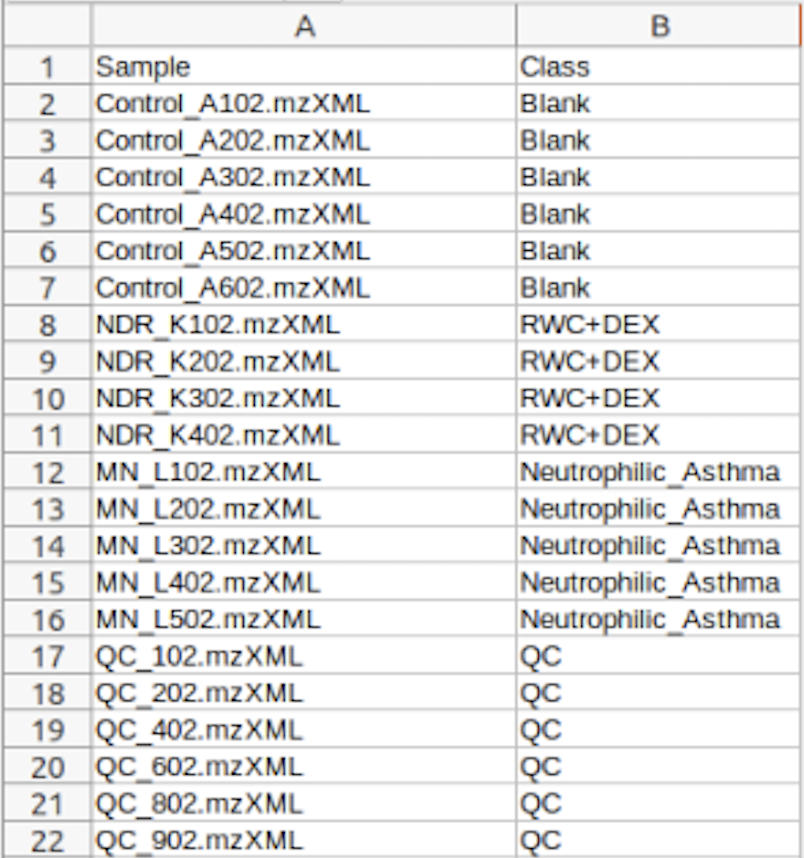
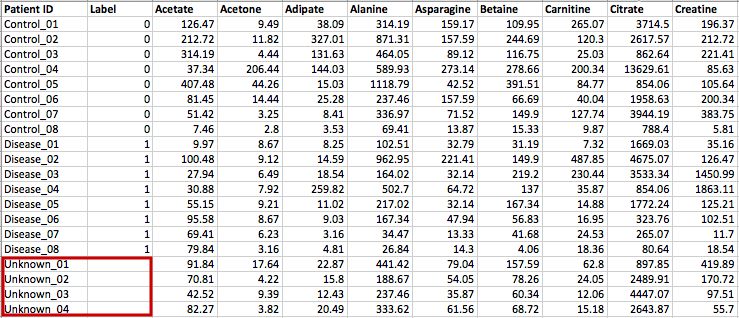
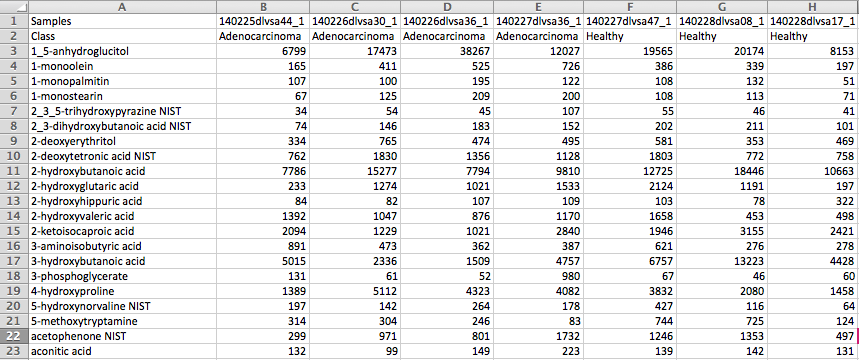
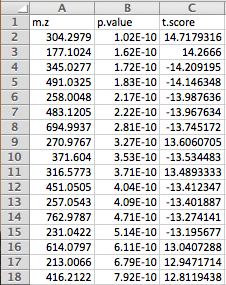
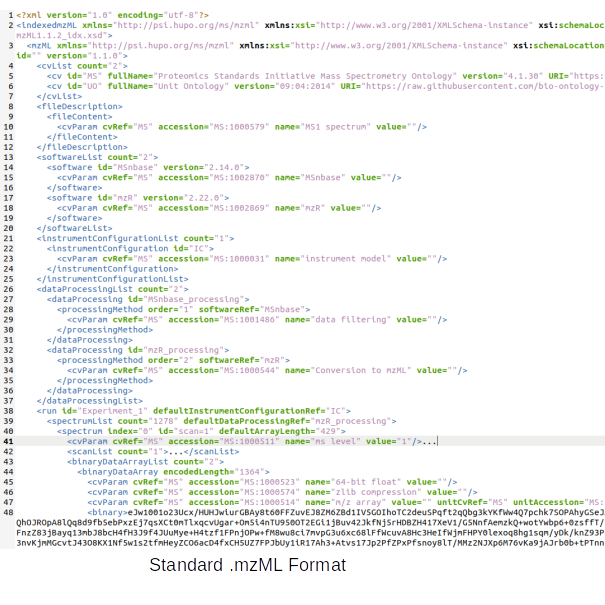
Data Formats:
Various example datasets available for different analysis purposes. You can download to inspect their formats, or scroll down for more detailed instructions.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
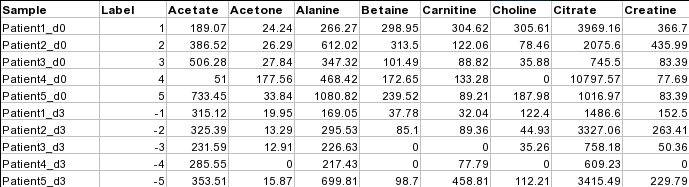
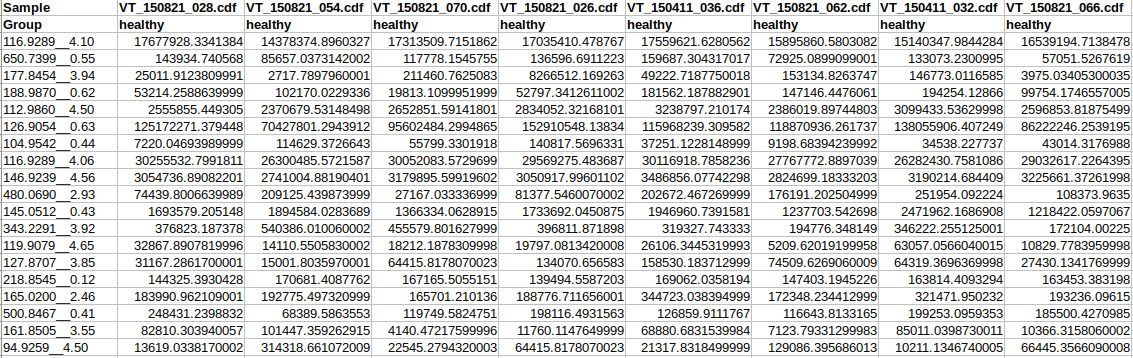
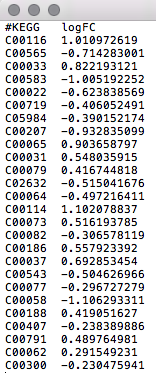
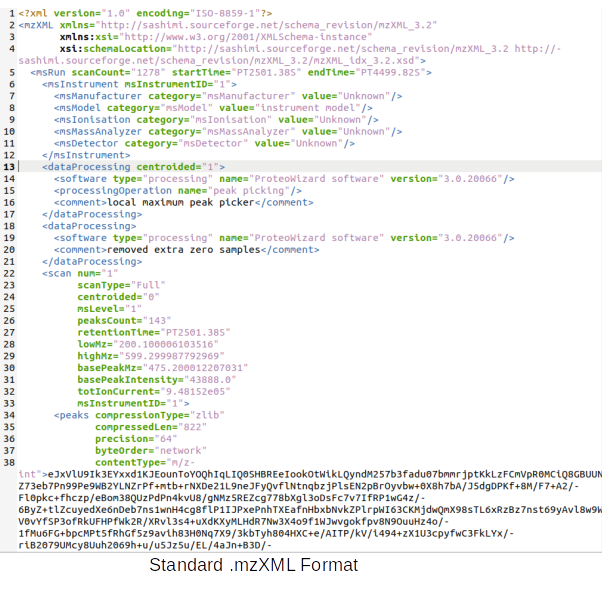
Data Formats:
Various example datasets available for different analysis purposes. You can download to inspect their formats, or scroll down for more detailed instructions.
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
{kind=link}
{kind=link}